

Researchers identify new dynamic mechanism of oxygen reduction reaction on single atom catalyst
Yang-Gang WANG 2022-04-12
In catalysis, oxygen reduction reaction (ORR) has been extensively studied for the purpose of developing renewable-energy technologies, such as fuel cells and metal-air batteries. However, due to the sluggish reaction kinetics, the ORR usually requires using highly efficient platinum-based catalysts, which are of high costs and scarce reserves.
To achieve the large-scale commercialization of the fuel cell technologies, a single atom catalyst based on transition metal dispersed N-doped nanocarbon is proposed to be a promising alternative for their low price, easy fabrication, and the potential of high electrochemical activity towards ORR. However, the underlying mechanism of ORR remains controversial due to the complexity of the electrochemical interface, which largely restricts the development and optimization of the single atom catalyst.
Associate Professor Yang-Gang Wang’s team from the Department of Chemistry at the Southern University of Science and Technology (SUSTech) recently published their findings on understanding the dynamic mechanism of oxygen reaction catalyzed by single atom catalyst dispersed on nitrogen-doped graphene.
Their research, entitled “Pseudo-adsorption and Long-range Redox Coupling during Oxygen Reduction Reaction on Single Atom Electrocatalyst,” was published online in Nature Communications, a multidisciplinary journal covering the natural sciences, including physics, chemistry, earth sciences, medicine, and biology.
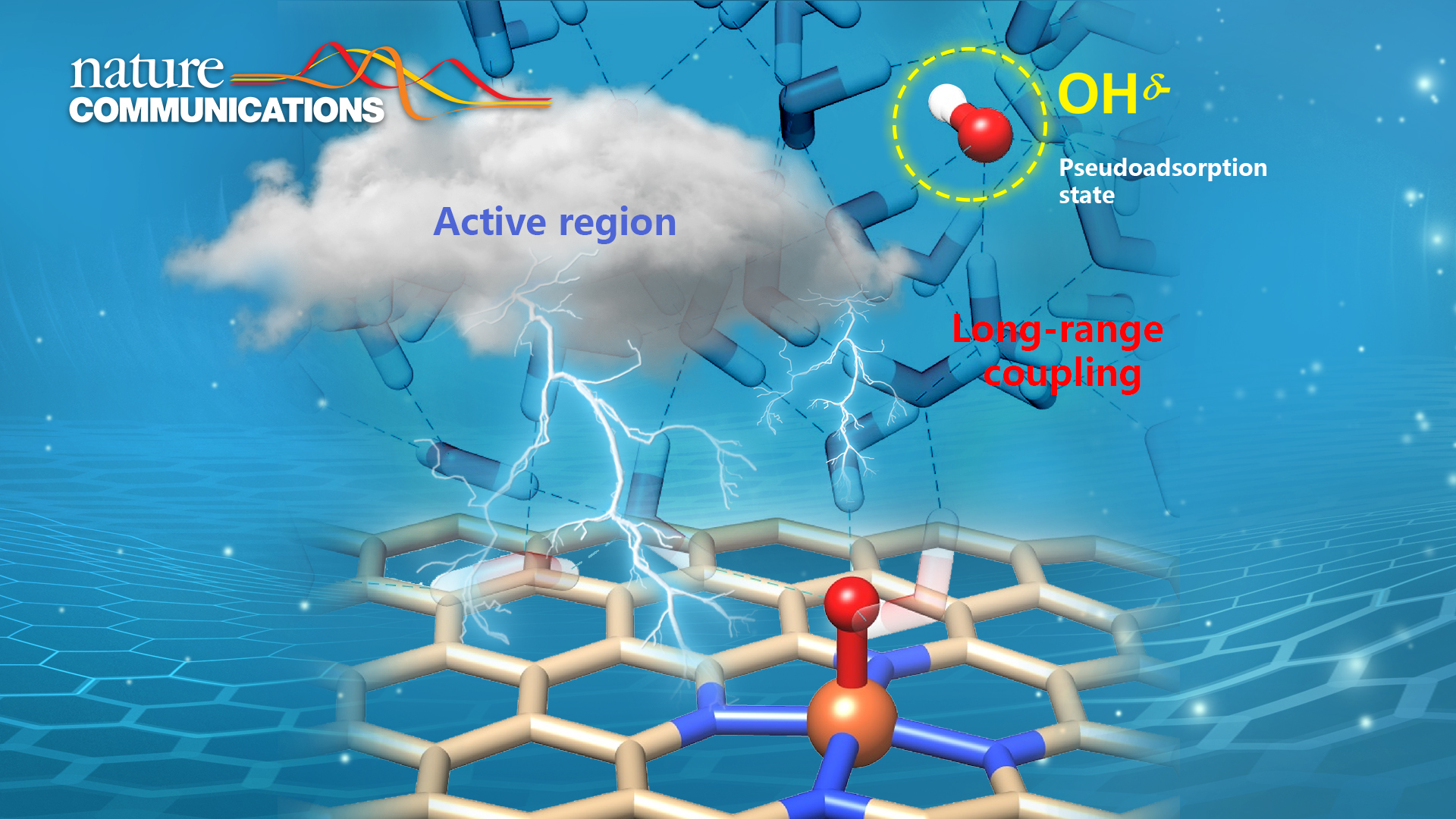
In this study, Prof. Wang’s team revisited the aqueous oxygen reduction reaction (ORR) mechanism on a series of transition metal (M=Fe, Co, Ni, Cu) single atom sites embedded in N-doped nanocarbon by ab initio molecular dynamics simulations and static calculations with explicit solvation. They identified the dissociative pathways and the thereby emerged solvated hydroxide species for all the proton-coupled electron transfer (PCET) steps at the electrochemical interface. Such hydroxide species can be dynamically confined in a “pseudo-adsorption” state at a few water layers away from the active site and respond to the redox event at the catalytic center in a coupled manner within a timescale less than 1 ps. In the PCET steps, the proton species (in the form of hydronium in neutral/acidic media or water in an alkaline medium) can protonate the pseudo-adsorbed hydroxide without needing to travel to the direct catalyst surface. Therefore, this expands the reactive region beyond the direct catalyst surface, boosting the reaction kinetics via alleviating mass transfer limits.
This work provides important implications for the catalysis community. In catalysis, the reaction species may not necessarily bind to the catalyst surface but be confined in an active region and long-range coupled with the active catalytic sites to complete the catalytic process. Understanding and harnessing the dynamic behaviors at different spatial scales in the electrochemical interface could unlock a new dimension for catalyst/reactor design and optimization.
Graduate students Jie-Wei Chen, Zisheng Zhang, and Hui-Min Yan from the Department of Chemistry at SUSTech are the co-first authors of this paper. Associate Prof. Yang-Gang Wang from the Department of Chemistry at SUSTech is the co-corresponding author.
This work was funded by the National Natural Science Foundation of China (NSFC), Guangdong Pearl River Talent Plan, and Guangdong Provincial Key Laboratory of Catalysis. The study was also supported by the Center for Computational Science and Engineering (CCSE) at SUSTech and the CHEM high-performance computing cluster (CHEM-HPC) located at the Department of Chemistry at SUSTech.
Paper link: https://doi.org/10.1038/s41467-022-29357-7